Yes, mitch can be used for pathway analysis of Methylation array data

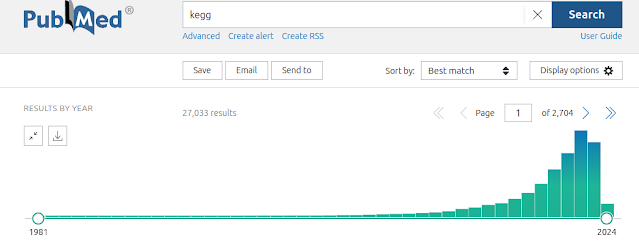
In 2020, Dr Antony Kaspi and I published a method called "mitch" [1] which is like GSEA, but was specifically designed for multi-contrast analysis, and based on rank-MANOVA statistics inspired by a 2012 paper by Cox and Mann. Mitch worked well for various types of omics data downstream of commonly used differential abundance tools like DESeq2, edgeR, DiffBind, etc, but we didn't consider at the time how mitch could be applied to microarray data. You might think that microarrays are outdated, but they are still used extensively for epigenome-wide association studies (EWASs), which are frequently used to understand disease processes and to identify biomarkers of disease. To demonstrate, there are 1536 publicly available methylation array studies on NCBI GEO, and probably many more thousands that are restricted access. The tools available for pathway analysis of methylation array data are a bit limited. There's an over-representation method that take into consideration