microRNA aligners compared
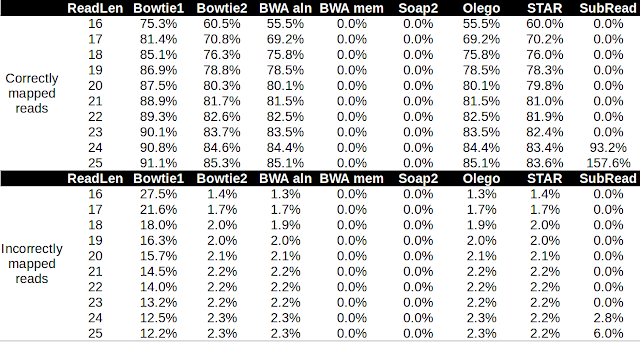
Alignment of microRNA to the genome poses a particular challenge because the reads are short, and some microRNAs are nearly identical. Moreover, microRNAs themselves are subject to RNA editing ( adenine-to-inosine conversion , non-templated base addition ) and normal sequencing error rates. In this post, I'm going to test the performance of several aligners in aligning microRNA reads to the Arabidopsis genome. I downloaded the Arabidopsis genome from Ensembl plant and the latest miRbase release version 21 . I used bowtie2 to align the 325 full-length hairpin transcripts to the Arabidopsis genome. I generated pseudo microRNA reads that uniformly cover the hairpin transcript at a range of lengths from 16 nt to 25 nt. I then aligned the reads to the Arabidopsis genome using these different aligners with the default settings. I then used bedtools and awk to count the correctly and incorrectly mapped reads at a mapQ threshold of 10. Table 1. Performance of several aligne
